The FDA has identified a list of Class I and Class II medical devices that are exempt from 510(k) and Good Manufacturing Practices (GMP) requirements, “subject to certain limitations.” If a device is exempt from 510(k) submission requirements, it may also be exempt from GMP requirements (but not the other way around). What do these two different scenarios mean for your organization and products?
Although FDA’s 510(k) program is the most commonly used device review pathway for medical devices in the US, not all devices need a formal submission to be legally marketed. Some medical devices are exempt from 510(k) clearance, meaning that manufacturers can market their device without clearance from the FDA. In addition, some of these exempt devices are also exempt from GMP requirements. What does this all mean? And is it going to save your company time and money?
You can determine whether a particular device is exempt from these requirements by searching the FDA’s Product Classification database.
In the following example, this product code is exempt from the 510(k) process, but still requires conformance to Good Manufacturing Practices:
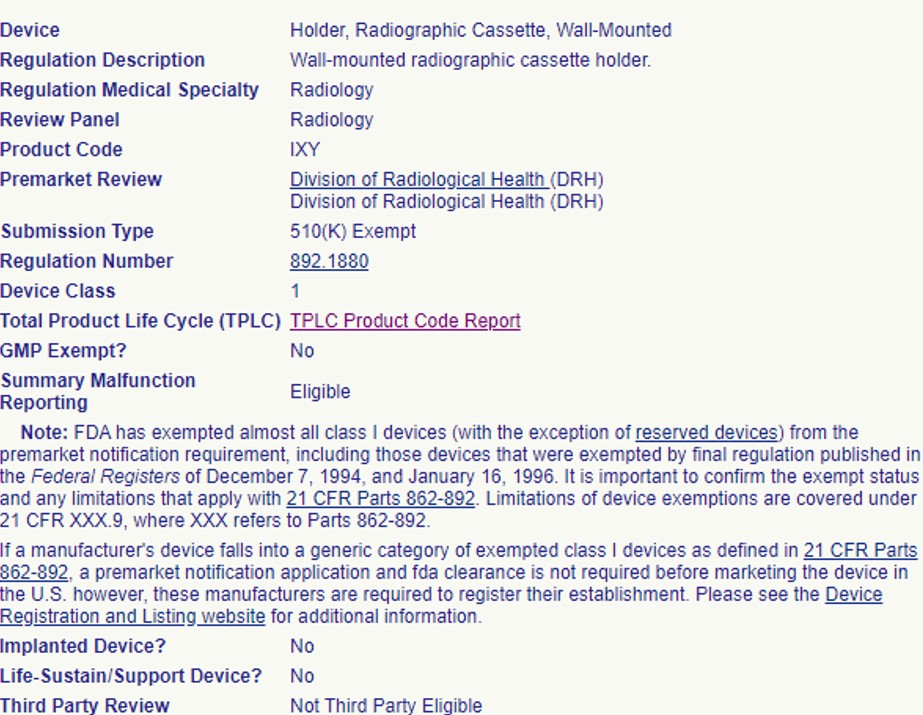
Some Class I and Class II devices are exempt from both the 510(k) process and GMP requirements (with some limitations). Others are just exempt from 510(k), but not GMP. Let’s explore each of these cases further:
510(k) Exempt Only (but not GMP exempt)
Most Class I devices and some Class II devices fall into this category. What does this mean?
- FDA still expects that Current Good Manufacturing Practices (GMP / cGMP) are followed, even though a 510(k) premarket notification submission is not required.
- You will still need to register your establishment and list your product(s) with the FDA
Most of these Class I devices do not require Design Controls (see device exceptions in 21 CFR 820.30). However, a Device Master Record (DMR) is required. We generally recommend that our customers try to conform to Design Controls, even if taking a “light touch,” because it provides additional assurance that the device is safe and effective, and allows fewer barriers to international market entry. We also recommend reviewing relevant standards and guidance for inclusion in the design process.
510K Exempt and GMP Exempt
Some Class I devices fall into this category:
- Products in this category are exempt from the GMP and QSR regulation, except for the requirements for maintaining records (21 CFR 820.180) and complaint files (21 CFR 820.198)
- Use caution: devices are not exempt from GMP if the device labeled or otherwise represented as sterile, or if the device contains software.
- You will still need to register your establishment and list your products with the FDA.
—
CMD can help your company strategize on the regulatory classification path of your device, including supporting discussions with the FDA and ensuring that time and effort is not spent complying with unnecessary regulations.